Wie entsteht Amyloidose?
Ablagerung von Transthyretin-Eiweiß
Der Auslöser der Erkrankung liegt in den Zellen des Körpers; genauer gesagt in den Leberzellen. Hier wird ein Eiweiß produziert, das im Körper normalerweise eine Transportfunktion übernimmt. Das betroffene Eiweiß heißt „Transthyretin“ (TTR). Es hat der Krankheit Transthyretin-Amyloidose den ersten Teil ihres Namens gegeben. Vier Transthyretin-Bausteine, sogenannte
„Monomere“, bilden zusammen einen Transportkörper – so wie vier Räder einen „Transportwagen“ bilden. Bei gesunden Menschen ist das Transthyretin für den Transport von Vitamin A und Schilddrüsenhormonen verantwortlich.
Bei Amyloidose-Patient:innen ist der „Transportwagen“ beschädigt:
Er ist nicht stabil und fällt auseinander. Die Einzelteile verklumpen und können ihre biologische Transportfunktion nicht mehr erfüllen. Für den Körper sind diese Eiweiße unbrauchbar geworden. Mediziner:innen sprechen von fehlgefalteten Eiweißen, die sich im Körper ablagern.
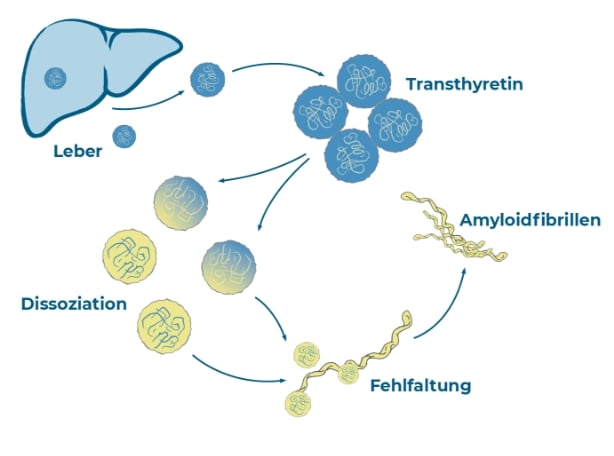
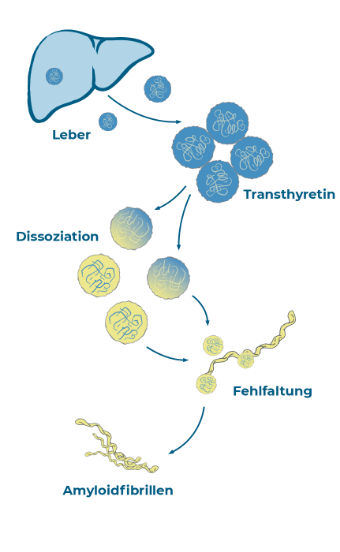
Der Transportwagen fällt auseinander, die Einzelteile verklumpen und bilden lange Fäden.
Was passiert mit den fehlgefalteten Eiweißen?
Die fehlgefalteten Eiweiße bilden lange Fäden. Diese Gebilde werden „Amyloid-Fibrillen“ genannt.
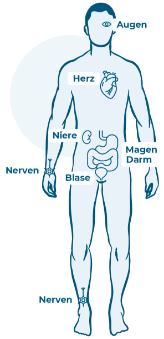
Die Eiweiße lagern sich an Organen oder Nerven an.
Das Problem: Die fadenförmigen Eiweißstrukturen kann der Körper nicht mehr weiterverarbeiten. Er kann sie weder in Einzelteile zerlegen, noch ausscheiden. Deshalb verklumpen die Eiweiße und lagern sich an unterschiedlichen Organen (z.B. Herz, Magen-Darm, Nieren oder Augen) oder an den Nerven an. Je mehr Eiweißablagerungen sich ansammeln, desto größer werden die Beschwerden, weil die Organe oder Nerven in ihrer Funktion beeinträchtigt werden.
Krankheiten, die durch die Ablagerungen von Amyloid-Fibrillen entstehen, heißen „Amyloidosen“.
Bei Amyloidose-Patient:innen ist der „Transportwagen“ beschädigt:
Je nachdem, wo sich die Eiweiße ablagern und zu Beschwerden führen, wird die Erkrankung in Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) oder Transthyretin-Amyloidose mit Polyneuropathie (ATTR-PN) unterschieden.
Warum ist der Transportwagen beschädigt?
ATTR-Amyloidose ohne genetische Ursache
Die nicht erbliche Form der ATTR-Amyloidose wird als Wildtyp ATTR-Amyloidose bezeichnet. In der Literatur spricht man von „wtATTR-Amyloidose“. Dabei kommt es ohne genetische Mutation, also ohne beschädigten Bauplan, zu Ablagerungen von Amyloidfibrillen in verschiedenen Organen. Bei einer Wildtyp-Erkrankung ist dabei besonders oft das Herz betroffen, wodurch es zur Ausprägung einer Transthyretin-Amyloidose mit Kardiomyopathie (ATTR-CM) kommt. Vorwiegend erkranken Männer über 65 Jahre. Die Ursache dieser Form der Erkrankung und damit der Grund für die Ablagerungen ist nicht vollständig bekannt. Eine Wildtyp ATTR-Amyloidose kann nicht vererbt werden.
ATTR-Amyloidose durch Mutation im Transthyretin-Gen
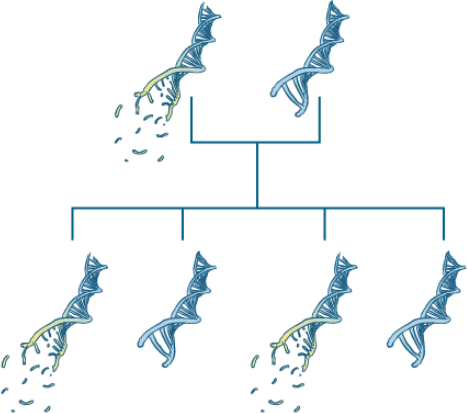
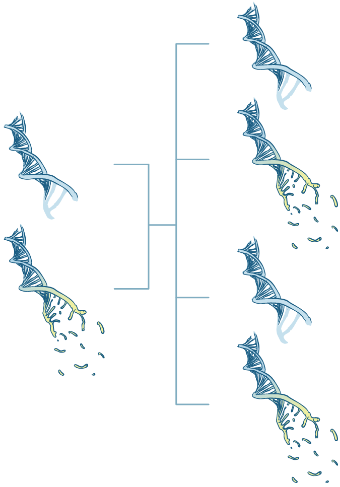
Eltern können den Gendefekt an ihre Kinder vererben.
Bei einem Teil der Patient:innen ist der beschädigte Bauplan für das Transthyretin-Protein vererbt. Medizinische Fachkreise sprechen dann von der „hereditären“ Form (vergl. engl. heritage = Erbe). Das kleine „h“ vor der Buchstabenkombination ATTR weist auf die angeborene Form der Erkrankung hin (hATTR-Amyloidose).
Bei dieser Form der ATTR-Amyloidose ist eine zufällige Genveränderung, eine sogenannte Mutation, des TTR-Gens für die Erkrankung verantwortlich. Weltweit sind über 120 verschiedene Mutationen bekannt, die zu einer Transthyretin-Amyloidose führen. und auch in Deutschland gibt es ein breites Spektrum an Mutationen. Je nachdem, welche Mutation bei den Betroffenen vorliegt, können sich die Symptome und der Schweregrad der ATTR-Amyloidose unterscheiden.
Mit einer geschätzten Krankheitshäufigkeit von 1:100.000 ist die erbliche Form der Transthyretin- Amyloidose eine sehr seltene Erkrankung.
Gendefekte können die ganze Familie treffen
Wird die ATTR-Amyloidose vererbt, ist die Erkrankung in der Familie häufig bereits bekannt. Dies kann bei der Diagnose der Amyloidose sehr hilfreich sein. Eltern, Großeltern oder andere Familienangehörige litten oder leiden an ähnlichen Symptomen. Menschen mit vererbter ATTR-Amyloidose produzieren von Geburt an fehlgebildetes Transthyretin-Protein. Die ersten Symptome der Ablagerungen zeigen sich jedoch meist erst im Erwachsenenalter, zwischen dem 25. und 35. Lebensjahr oder dem 55. und 65. Lebensjahr.